 药学进展
药学进展专家介绍:朱继东

2003 年毕业于中国科学院上海有机化学研究所,并获有机化学博士学位,2003—2008年在美国哈佛大学医学院细胞生物学系从事博士后研究。2008 年归国后,在诺华制药公司担任药物研发实验室研究员、主任,从事表观遗传学靶点药物的开发工作。于 2014 年加入中国科学院上海有机化学研究所,任“百人计划”研究员、博士生导师、课题组组长。主要工作集中在“不可靶向”抗肿瘤靶点的小分子抑制剂开发和新型肿瘤免疫药物的开发等,并取得了重大进展;主持和参与了科技部“重点研发计划”、基金委“重点项目”、中科院“先导科技专项”等多项国家重大课题。研究成果作为通讯作者论文发表在 Proc Natl AcadSci USA、J Med Chem 等国际学术期刊上,并申请多项专利,部分成果已成功实现转化。
正文
蛋白磷酸酶变构抑制剂的研究进展
曹恒义,朱继东 *
(中国科学院上海有机化学研究所生物与化学交叉研究中心,上海 201203)
[摘要]蛋白磷酸酶是一类以蛋白质为底物使其去磷酸化的酶,与蛋白激酶的磷酸化作用刚好相反,二者共同调节生物体内各种蛋白质的磷酸化水平,在生物信号传递中发挥着重要的作用。最近的研究表明,一些蛋白磷酸酶的功能异常与肿瘤、糖尿病和自身免疫性疾病等都有着密切的关联。不同于蛋白激酶的研究,目前对于蛋白磷酸酶在体内调控机制的理解尚浅,且缺乏高活性和特异性的小分子蛋白磷酸酶抑制剂作为化学探针,因此以蛋白磷酸酶为靶标开发治疗药物的研究一直进展缓慢。近年来,一系列作用于蛋白磷酸酶催化位点以外的小分子变构抑制剂逐渐被发现,为蛋白磷酸酶的研究带来新的进展,综述这些新的蛋白磷酸酶变构抑制剂的研究进展,重点介绍与疾病相关且作用位点明确的小分子抑制剂,简要介绍其发现过程,以期为更多的蛋白磷酸酶变构抑制剂的研究工作带来启发。
生物体内的磷酸化调控是调节细胞生命活动的最重要的调控方式之一。其广泛地发生在蛋白、磷脂、糖类和核糖核苷酸等诸多细胞成分中。蛋白激酶和蛋白磷酸酶分别作为磷酸化和去磷酸化功能的两大类蛋白,在细胞中维系着蛋白磷酸化调控的平衡,因此对它们功能的研究显得极为重要。蛋白质的磷酸化一般发生在丝氨酸、苏氨酸和酪氨酸上,蛋白激酶通常以 ATP 为底物将磷酸基团转移到这些氨基酸残基上使其磷酸化,从而调节蛋白的功能或稳定性,或者将细胞信号继续传递下去,蛋白磷酸酶则会将蛋白氨基酸残基上的磷酸水解。
目前发现 107 个蛋白磷酸酶亚家族,可以分为如下 4 类:蛋白酪氨酸磷酸酶(protein tyrosinephosphatases,PTPs)、金属离子依赖性蛋白磷酸酶(metal-dependent protein phosphatases,PPMs)、磷蛋白磷酸酶(phosphoprotein phosphatases,PPPs)以及卤酸脱卤型磷酸酶(phosphatases of the haloaciddehalogenases,HAD 磷酸酶),其中 PPMs 和 PPPs属于蛋白丝 / 苏氨酸磷酸酶,HAD 磷酸酶中除了缺眼蛋白家族(eyes absent family of proteins,Eya)属于酪氨酸磷酸酶以外,其余已知的均为丝/苏氨酸磷酸酶。PTPs 根据催化模组的不同又分为第 1、2、3 类 PTPs 以及细菌 PTPs。第 1 类 PTPs 包括受体型 PTPs、非受体型 PTPs 和双特异性磷酸酶(dualspecificity phosphatases,DSPs),DSPs 既可以使酪氨酸去磷酸化,也可以使丝/苏氨酸去磷酸化。
由于蛋白激酶的激活功能往往与肿瘤等疾病直接相关,因此蛋白激酶很早就被作为药物靶点来进行研究,其抑制剂也已进入临床应用,然而,蛋白磷酸酶抑制剂的研究相对较少。作为蛋白激酶的主要拮抗蛋白,蛋白磷酸酶一开始被简单地认为是肿瘤抑制因子,但后来随着研究的深入,人们发现一些蛋白磷酸酶本身可能就是致癌因子,并且一些导致蛋白磷酸酶持续激活的基因异常已被证实在肿瘤等疾病中广泛存在,是重要的原癌基因。随着对蛋白磷酸酶去磷酸化作用的研究继续深入,蛋白磷酸酶抑制剂的潜在临床治疗价值得到越来越多的实验验证;同时,对蛋白磷酸酶活性有调节作用的小分子也为致病机制研究提供了重要的工具。早期有一些天然产物被发现具有蛋白磷酸酶抑制能力,但它们均主要在两个方面存在一些问题而最终无法进入临床应用:一是特异性问题,这些抑制剂多作用于磷酸酶的催化域,而同家族蛋白磷酸酶催化域高度保守,抑制剂难以区分不同亚型的磷酸酶,从而带来潜在的副作用,并给药理机制研究造成困难;二是蛋白磷酸酶的催化域往往带有正电荷,更倾向于结合带负电的分子,而带负电荷的小分子抑制剂本身在代谢和生物利用度方面处于弱势。最早,人们设计蛋白磷酸酶底物类似物来竞争蛋白磷酸酶的作用,但绝大多数都因成药性差而失败。直到最近,一些分子通过作用于蛋白磷酸酶催化域以外的潜在口袋来调节蛋白磷酸酶的活性(见表 1),巧妙地回避了蛋白磷酸酶催化域抑制剂特异性差和成药性差的问题,这些新的蛋白磷酸酶变构抑制剂的问世为蛋白磷酸酶的研究带来新的活力,也让蛋白磷酸酶作为药物靶点重获希望。本文就目前热门的蛋白磷酸酶变构抑制剂的研究进展作一综述,希望为更多新的蛋白磷酸酶变构抑制剂研究带来启发。
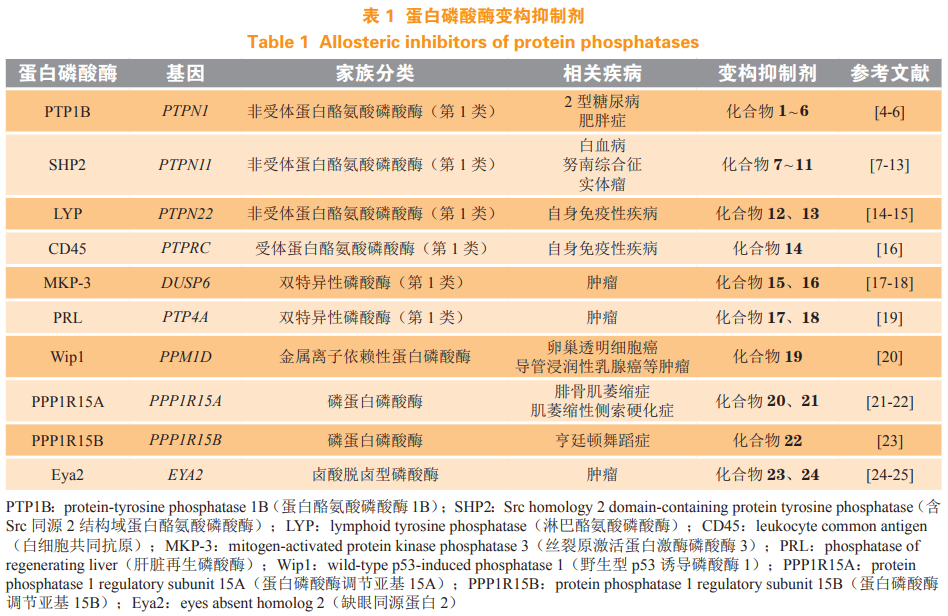
1 蛋白酪氨酸磷酸酶 1B 变构抑制剂
蛋白酪氨酸磷酸酶 1B (protein-tyrosine phosphatase1B,PTP1B)是由 PTPN1 基因编码的一种非受体蛋白酪氨酸磷酸酶(non-receptor PTP)。越来越多的证据表明,PTP1B 在多种肿瘤中都存在高表达,如前列腺癌、上皮癌、卵巢癌、乳腺癌和胃肠道癌等,因此有人将其视为能够促进肿瘤生长的蛋白磷酸酶。更让人感兴趣的是,PTP1B具有减弱细胞对胰岛素敏感性的作用,其可以使胰岛素受体 b 基(insulin receptor b subunit,IRb)的 pY1162/pY1163 酪氨酸以及胰岛素受体底物-1(insulin receptor substrate-1,IRS-1)去磷酸化从而减弱胰岛素诱导的蛋白激酶 B(protein kinase B,PKB/Akt)磷酸化,当 PTP1B 被抑制后可恢复细胞对胰岛素的敏感性,因此 PTP1B 的抑制剂有望成为治疗 2 型糖尿病的新方案。此外,PTP1B 对瘦素(leptin)信号通路也有一定的作用,PTP1B 抑制剂也被开发用于肥胖症的治疗。
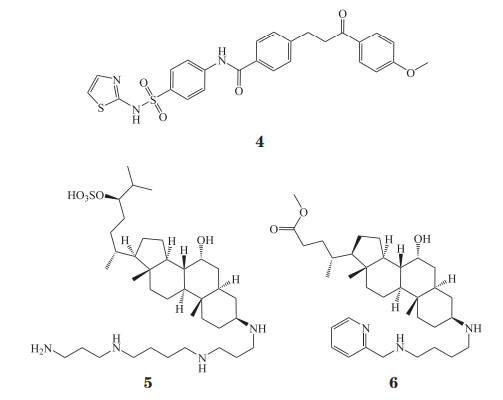
文献报道的 PTP1B 抑制剂已有很多,既有天然产物也有人工合成的小分子抑制剂,它们绝大多数都作用在催化位点上,虽然在体外试验中具有抑制活性,但在选择性和细胞渗透性上仍然存在很大的局限性,因此开发催化位点之外的抑制剂是非常有希望突破这种局限的方案。1997 年,Puius等在催化位点附近发现了芳基磷酸结合位点,这一位点的氨基酸残基序列相较于催化位点在同源蛋白中具有更大的差异,随后越来越多的 Cys215 残基附近的位点被发现,针对这些位点和催化位点设计的双位点抑制剂成功解决了一部分蛋白磷酸酶抑制剂选择性差的问题,但由于这些抑制剂仍然依赖催化位点的结合因而仍具有带电荷基团或易被代谢的基团,不利于成药。2004 年,Wiesmann 等报道了首个 PTP1B 变构抑制剂,他们通过酶动力学试验测试了一些非磷酸结构的 PTP1B 抑制剂及衍生物,从中发现了一个不与底物相竞争的化合物 1(IC50=350 µmol·L-1),随后改造的化合物 2(IC50=22 µmol· L-1)和 3(IC50 = 8 µmol· L-1)的抑制活性提高了约 40 倍,继而通过共晶结构发现化合物 2 结合在新的变构位点上,该位点位于 α3 螺旋和 α6 螺旋之间的凹槽,抑制剂作用于这一位点可以阻止对催化区域十分重要的WPD loop关闭构象(活性构象)的形成(见图 1)。由于化合物 2 作用在同源性很高的催化区域之外,随后的选择性试验也发现它对同源蛋白 LAR(IC50 >500 µmol·L-1)和 TC-PTP(IC50=129 µmol·L-1)的抑制活性与 PTP1B 存在明显差异。细胞试验也证实了这些化合物具有增强胰岛素信号传导的作用,这一发现极大地激发了研究人员对蛋白磷酸酶抑制剂研发的热情。Tang 等后来在化合物 1~3 的结构基础上进一步改造得到化合物 4(IC50=3.2 µmol·L-1)。2014 年,Krishnan 等通过靶点筛选发现 MSI-1436(5,又名 trodusquemine)可作用于 PTP1B(对 PTP1B1-405 的 IC50 为 600 nmol·L-1),并结合磁共振(NMR)和蛋白晶体数据分析了更长片段的 PTP1B 蛋白的变构位点的作用机制,发现 MSI-1436 可以作用在一个新的变构位点,该位点位于 C 端的 α9' 螺旋上。小鼠移植瘤模型实验发现,MSI-1436 能抑制人表皮生长因子受体-2(humanepidermal growth factor receptor 2,HER2)阳性的乳腺癌细胞生长。遗憾的是,由于 MSI-1436 活性较低且在体内的生物利用度较差,DepYmed 公司于 2017 年停止了该化合物的临床试验,目前,其改造后的第 2 代分子 DPM-1001(6,对 PTP1B1-405 的IC50 为 100 nmol·L-1)正在进行临床前研究,该分子可以与 Cu2+ 形成稳定的配合物,并且该配合物可能与 C 末端的组氨酸结合。


2 含 Src 同源 2 结构域蛋白酪氨酸磷酸酶变构 抑制剂
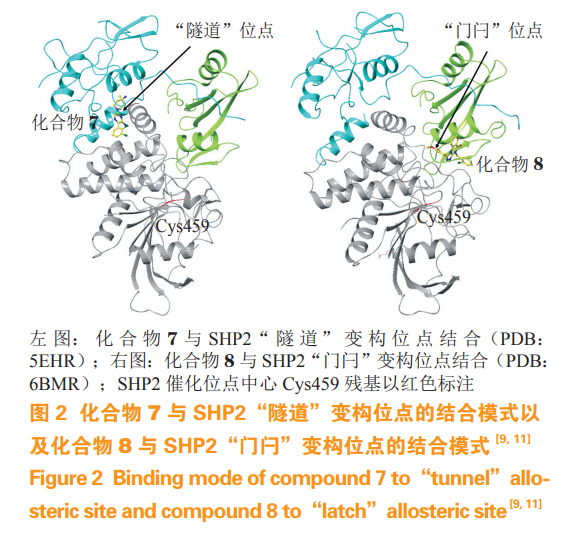
含 Src 同源 2 结构域蛋白酪氨酸磷酸酶(Srchomology 2 domain containing protein tyrosinephosphatase,SHP2)由 PTPN11 基因编码,其结构包含 N 端的 2 个 Src 同源 2 结构域(N-SH2 和 C-SH2domains)和 C 端的 1 个具有催化功能的 PTP 结构域。在通常状态下,N 端的 SH2 结构域会与 PTP 结构域相互作用形成一种自抑制的构象,当 SH2 结构域与酪氨酸磷酸化的生长因子受体结合后会解除SHP2 的自抑制构象,形成开放的活化状态从而激活PTP 结构域的去磷酸化功能。活化的 SHP2 可以激活 RAS- 细胞外调节蛋白激酶(extracellular signalregulated kinase,ERK) 信 号通路,因 此 SHP2 是诸多蛋白激酶通路上的重要调控蛋白。最近也有研究表明,SHP2 可以通过结合程序性死亡受体 1(programmed cell death protein 1,PD-1)和 B、T 淋巴细胞衰减因子(B- and T-lymphocyte attenuator,BTLA)等抑制性的免疫检查点来调节 T 细胞的活化。同时,PTPN11 基因的突变导致 SHP2 蛋白的持续激活在多种人类发育性疾病和癌症中都被发现,如白血、努南综合征等,因此被认定为是原癌基因。早期针对其 PTP 结构域的抑制剂同样存在选择性差、活性低、成药性差等缺点。2016年,诺华(Novartis)公司报道了化合物 SHP099(7,IC50 = 0.071 µmol· L-1),该化合物不同于以往的 SHP2 抑制剂,共晶研究发现它能结合到 2 个SH2 结构域和 PTP 结构域三者之间(称作“隧道”位点),使 SHP2 稳定在关闭的自抑制构象上(见图 2),从而抑制 SHP2 的活性。这一独特的作用方式使 SHP099 成为首个高效、可口服、选择性好的 SHP2 抑制剂。在随后的研究中,Fodor 等又发现了 SHP2 结构上的另一个变构位点 ——“门闩”位点,该位点位于 N-SH2 结构域和 PTP 结构域交界面之间,同样可以稳定 SHP2 的自抑制构象(见图 2),针对该位点筛选得到变构抑制剂 SHP244(8,对 SHP21-525 的 IC50 为 60 µmol· L-1),其作为苗头化合物经进一步改造后,抑制活性有所提升,但该位点的抑制剂活性相较作用于“隧道”位点的抑制剂仍然较弱,尽管如此,新的位点也具有很好的开发潜力,对未来解决可能出现的变构位点突变产生耐药的情况有所帮助。最近,诺华公司的 SHP2 变构抑制剂开发又有了新的进展,在接连发表的 2 篇文章中,诺华公司对 SHP099 进行了进一步改造,进一步扩充了“隧道”位点变构抑制剂的化合物库,开发了一系列活性更高、药物代谢性质更好的化合物,目前 TNO155(9)已处于Ⅰ期临床试验中,这充分说明了磷酸酶的变构抑制剂具有很大的药物开发潜力。在 SHP099 被报道之后,越来越多的研究机构和医药企业开始致力于 SHP2 变构抑制剂的研发,SHP2 也成为明星靶点。Xie 等在 SHP2 的“隧道”位点关键位置引入与疾病相关的 E76A 突变,利用 SHP2E76A 持续激活的蛋白来筛选有效的变构抑制剂,发现经过改造得到的化合物 10(对 SHP2E76A 的IC50 为 0.71 µmol·L-1)能有效抑制丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路。Nichols 等发现 SHP099 的类似物 RMC-4550(11,IC50 =0.58 nmol·L-1)可以对 BRAF 第 3 类突变、神经纤维瘤蛋白 1(neurofibromin 1,NF1)缺失以及KRAS(G12C) 等依赖 GTP 激活 RAS-MAPK 通路的癌症驱动基因产生抑制,目前 Revolution Medicines公司的 RMC-4630 也已处于Ⅰ/Ⅱ期临床试验阶段。

3 淋巴酪氨酸磷酸酶变构抑制剂
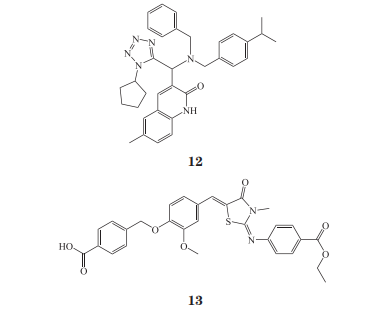
淋巴酪氨酸磷酸酶(lymphoid tyrosine phosphatase,LYP)是由 PTPN22 基因编码的磷酸酶,催化位点中心在 Cys227,它在 T 细胞信号调节中有着重要的作用并有可能成为治疗自身免疫性疾病的靶点。LYPR620W 杂合突变在 1 型糖尿病、类风湿性关节炎、格雷夫斯病以及一些自身免疫性疾病中均有发现,但是其发病机制尚不十分明确,近期的研究表明,突变的 LYP 可能导致 T 细胞整合素下游信号分子的磷酸化水平升高 。该靶点目前鲜有变构抑制剂的报道,Stanford 等报道的 LYP 抑制剂不带有羧酸、磷酸等基团,他们通过动力学分析、多肽氨基酸氢/氘交换质谱分析[peptide amide hydrogen/deuterium exchange massspectrometry (DXMS) analysis]、分子对接(docking)以及对关键氨基酸残基进行突变,发现他们的抑制剂 12(IC50 = 5.28 µmol · L-1)作用于催化位点之外的位点,因此具有良好的选择性。最近,Li 等发现了又一 LYP 变构抑制剂 NC1(13,Ki = 4.3μmol · L-1),他们运用非天然氨基酸 2-氟-酪氨酸(2-fluorine-tyrosine,F2Y)替换 Leu281,并用19F-NMR 观测 WPD loop 的运动,发现该抑制剂能限制 WPD loop 的构象。
4 白细胞共同抗原变构抑制剂
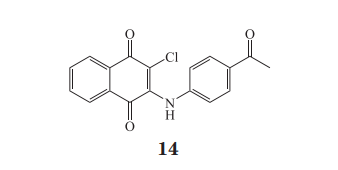
白细胞共同抗原(leukocyte common antigen,即 CD45)是一种在白细胞膜上表达的受体蛋白酪氨酸磷酸酶,由 PTPRC 基因编码,其功能对 T 细胞受体(T-cell receptor,TCR)的信号转导以及 T细胞的激活至关重要,它可以调节 Src 家族蛋白激酶的淋巴细胞特异性酪氨酸蛋白激酶(lymphocytespecific protein tyrosine kinase,Lck)等,将其 pY505去磷酸化(待激活的活化态,“primed” status)从而促进 Lck pY394(小鼠 pY393)的自磷酸化激活;此外,pY394 的去磷酸化也受到高浓度 CD45的调节,因此 CD45 作为双向调节 Lck 等蛋白活性的关键因子,维持着 Lck 等蛋白的磷酸化平衡,避免 Lck 持续的超活化状态。CD45 由胞外糖基化的可变结构域、1 个单次跨膜结构域以及保守的胞内的 D1、D2 结构域组成,其中 D1 结构域具有催化磷酸基团水解的半胱氨酸残基,D2 则不具有催化活性。当 PTPRC 基因发生改变以后,会导致可变剪接发生变化使 CD45 表达的亚型发生改变,这与多种自身免疫性疾病相关,如多发性硬化症、系统性硬化症、系统性红斑狼疮、自身免疫性肝炎等,因此 CD45 也被视为十分有价值的治疗自身免疫性疾病的靶点。此外,CD45 表达水平下降也被发现在对抗埃博拉病毒和炭疽感染中具有一定的作用。Perron 等先通过虚拟筛选得到了可能作用在 D1、D2 结构域之间的化合物,从中发现了一个选择性较好的苗头化合物,进一步改造得到化合物 14(IC50 = 200 nmol · L-1)并在突变试验中验证了其结合位点,作者推测其可能通过该变构位点影响 CD45 的构象和活性,使 Lck 的pY393 增加,活化态的 Lck 减少,阻断 TCR 信号,迟发性过敏小鼠模型实验表明,该化合物可减少炎症反应。
5 丝裂原激活蛋白激酶磷酸酶 3 变构抑制剂
丝裂原激活蛋白激酶磷酸酶 3(mitogen-activatedprotein kinase phosphatase 3,MKP-3)又称为 DUSP6,是一个广泛存在的 DSP 家族的蛋白磷酸酶,除了解除酪氨酸的磷酸化,还可以对 MAPK、ERK 等的丝/苏氨酸去磷酸化,其功能主要是负反馈调节成纤维细胞生长因子(fibroblast growth factor,FGF)信号。MKP-3 作为 RAS-ERK 通路的抑制蛋白,其抑制剂目前主要被用于一些胚胎发育、免疫和 FGF 相关的研究。尽管如此,也有一些文献报道 MKP-3 可能促进肿瘤的产生。目前 MKP-3 变构抑制剂很少,Molina 研究团队通过转基因斑马鱼构建了体内筛选模型,最终发现 MKP-3 抑制剂 BCI(15,斑马鱼 FGFR 激活试验 EC50 为 10.6 µmol· L-1),该抑制剂能有效抑制 ERK 介导的 MKP-3 活化,从而抑制FGF 信号,并发现其可能作用于催化位点旁的变构位点上,随后改造的 BCI-215(16,EC50= 12.0 µmol·L-1)相较于 BCI 有着更低的细胞毒性。2017年,Kaltenmeier 等发现 BCI-215 可以在 MDABN-231 人乳腺癌细胞中选择性激活 MAPK 信号通路引起细胞坏死。

6 肝脏再生磷酸酶变构抑制剂
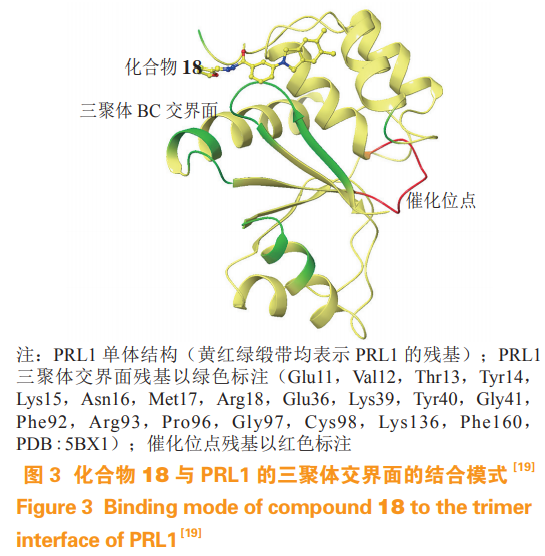
肝脏再生磷酸酶(phosphatases of regeneratingliver,PRL)主要有 PRL-1、PRL-2 和 PRL-3 等 3个亚型,分别由 PTP4A1、PTP4A2、PTP4A3 基因编码,三者基因序列高度相似。有证据表明PRL 在促进肿瘤转移侵袭过程中有重要的作用,并在多种肿瘤中高表达。由于 PRL 催化区十分平坦,没有明显的小分子结合口袋,并且与其他 PTP家族蛋白的催化区高度相似,因此给 PRL 选择性抑制剂的设计带来挑战。值得庆幸的是,PRL 蛋白在细胞内需要通过相互结合形成同源三聚体才能发挥促进细胞迁移的功能,Bai 等根据这一特点,通过虚拟筛选的方法发现了作用在三聚体交界面的变构抑制剂 17,其在 10 µmol· L-1 浓度下可抑制PRL1 过表达细胞增殖和迁移,能有效地在体外和小鼠移植瘤模型中抑制肿瘤生长;在共晶结构中,其类似物 Analog-3(18)与 PRL1 单体结合在三聚体的交界面上(见图 3)。
7 野生型 p53 诱导磷酸酶变构抑制剂
野生型 p53 诱导磷酸酶 1(wild-type p53-inducedphosphatase 1,Wip1)是由 PPM1D 基因编码的丝/苏氨酸磷酸酶,可以负向调节 DNA 损伤应答通路(DNA damage response pathway,DDR)上的关键蛋白,如 p53 等。当细胞因电离辐射、化疗药物而造成损伤时,细胞会启动 DNA 损伤应答通路来减缓细胞周期,修复 DNA,或启动细胞凋亡,从而维持基因组的稳定。当损伤修复结束后,细胞则通过蛋白磷酸酶停止 DDR,其中 PP2C 丝 / 苏氨酸磷酸酶家族在其中发挥着重要作用,Wip1 就是由 p53诱导产生的这一类磷酸酶。目前在多种肿瘤中发现 PPM1D 基因扩增和 Wip1 过表达,因而 Wip1 抑制剂被认为能间接激活野生型 p53。
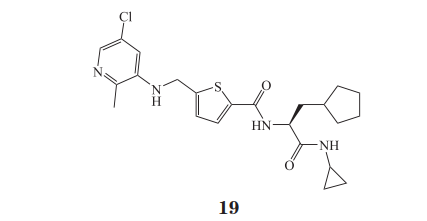
由于 Wip1 的结构仍未完全解析出来,目前唯一报道的作用于催化位点之外的抑制剂是 GSK2830371(19,对 Wip12-420 的 IC50 为 6 nmol·L-1)。Gilmartin等通过酶活性试验(以荧光素二磷酸酯为底物)和酶亲和力试验(利用 DNA 编码化合物库),分别采用生物化学和生物物理方法得到具有相似性的CAA(capped amino acid)小分子结构,经改造后得到 GSK2830371;由于未能成功得到共晶结构,他们采用了光亲和标记试验来验证 CAA 分子的结合位点,再结合同源蛋白 PPM1A 的结构分析,发现 CAA 分子结合在 Wip1 催化位点外的一段可翻动的“flap”片段附近,该片段对催化有着重要作用,因此该抑制剂具有很好的特异性,并且在小鼠移植瘤模型中也能有效抑制 B 细胞淋巴瘤的生长。
8 蛋白磷酸酶调节亚基 15A/15B 变构抑制剂
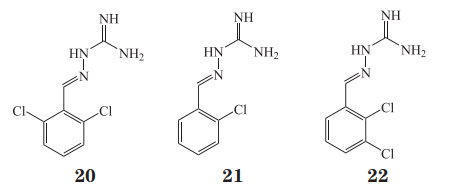
蛋白磷酸酶调节亚基 15A(protein phosphatase1 regulatory subunit 15A,PPP1R15A)和蛋白磷酸酶调节亚基 15B(protein phosphatase 1 regulatorysubunit 15B,PPP1R15B)是分别由 PPP1R15A 和PPP1R15B 编码的另一类蛋白丝/苏氨酸磷酸酶 ——PPPs,这一亚家族的结构与 PPMs 存在明显的差异, 由保守的催化亚 基(catalytic subunit proteinphosphatase 1,PP1c)和不同的调节亚基(如 R15A或 R15B)相结合而组成,众多的调节亚基决定了全酶的细胞定位以及底物特异性。最早,Tsaytler等发现了 α2 肾上腺素受体激动剂胍那苄(20,guanabenz,R15A-PP1c SPR试验:KD =0.122 μmol·L-1)可以选择性地结合在 PPP1R15A 上,干扰真核翻译起始因子 2-α 亚基(eukaryotic translation initiationfactor 2 subunit alpha,eIF2-α)的去磷酸化,增加磷酸化 eIF2-α 的水平可以短暂减缓蛋白质的合成,有利于分子伴侣对蛋白质正确折叠,维持蛋白质稳态。随后,该团队的 Das 等进一步开发了 PPP1R15A选择性抑制剂 sephin1(21,又名 IFB-088,KD =0.786 μmol· L-1),并在小鼠中预防了 2 种蛋白错误折叠相关疾病 —— 腓骨肌萎缩症(charcot-marietooth disease)和肌萎缩性侧索硬化症(amyotrophic lateral sclerosis)。2018 年,Krzyzosiak 等报道了在体外构建的一种以表面等离子共振(surfaceplasmon resonance,SPR)技术为基础的筛选平台,使用更为完整的 PP1c-R15A/B 蛋白来测试小分子和酶之间的亲和力,并筛选得到 PPP1R15B 选择性抑制剂 raphin1(22,R15B-PP1c SPR 试验:KD =0.033μmol· L-1),该化合物可口服且可跨越血脑屏障,在亨廷顿舞蹈症(Huntington’s disease)小鼠模型中证实对疾病有潜在的预防作用。
9 缺眼同源蛋白 2 变构抑制剂
Eya 最早被发现是作为转录因子 Six1 的共激活因子,二者在胚胎发育和细胞增殖中有重要的作用,正常成人组织的 Eya 几乎不表达,但在肿瘤中发现Six1 和 Eya 复合物的高表达。高表达的 Six1 和缺眼同源蛋白 2(eyes absent homolog 2,Eya2)可以增加肿瘤的侵袭性,缩短患者的生存期。目前发现的可以被 Eya 去磷酸化的蛋白有组蛋白H2AX、雌激素受体 β(estrogen receptor β,ER-β)和含 WD 重复结构域蛋白 1(WD repeat-containingprotein 1,WDR1),因此人们将 Eya 视为潜在的抗肿瘤药物靶点基于以下几种可能:当 DNA 受到损伤时,Eya 的磷酸酶活性可以使组蛋白 H2AX 去磷酸化,从而使细胞进行DNA修复而不进入凋亡,使用 Eya 抑制剂可能有助于增加患者对放化疗的敏感性;磷酸化的 ER-β 具有一定的抑制肿瘤生长的作用,高表达 Eya 会使 ER-β 失去这一作用,Eya抑制剂可能可以恢复 ER-β 的磷酸化水平;WDR1则与细胞骨架相关,可能影响细胞的侵袭性,但具体机制尚不明确。
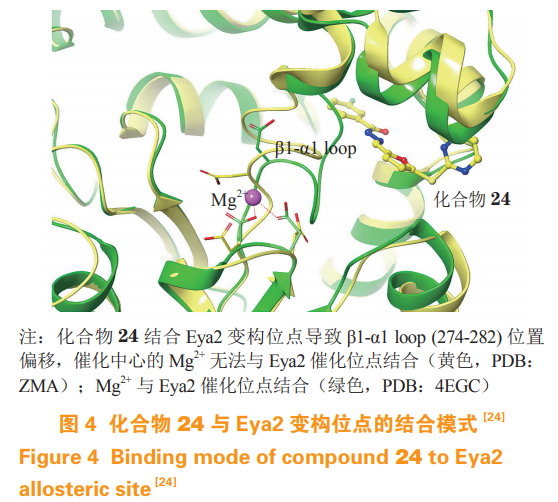
Eya 是一类经典的非巯基催化的蛋白磷酸酶(non-thiol-based protein phosphatases),属于卤酸脱卤酶家族,该家族通过天冬氨酸而非半胱氨酸催化去磷酸化。Krueger 等通过高通量筛选发现一类可以选择性抑制 Eya2 的化合物,其中一些化合物在高浓度 Mg2+ 下仍具有非竞争性的酶动力学特征,随后突变试验发现化合物 MLS000544460(23,对 Eya2253-538 的 IC50 为 4.1 µmol· L-1)可能并不作用在催化位点上。最近,该团队合成了溶解性更好的类似物 NCG00249987(24,对 Eya2253-538 的 IC50 为 3.0µmol·L-1),并通过晶体结构验证了其变构结合位点,占据该变构位点会导致催化位点的 β1-α1 loop(274-282)位置偏移,使得 Mg2+ 难以与催化位点结合(见图 4),从而抑制 Eya2 的活性,降低肿瘤细胞的侵袭性。

10 结论和展望
随着对蛋白磷酸酶研究的深入,人们更加深刻地认识到蛋白磷酸酶和蛋白激酶之间维系的磷酸化平衡在疾病中的重要作用,蛋白磷酸酶也不仅仅只发挥下调磷酸化信号的作用,异常的蛋白磷酸酶在不同环境下上调或下调磷酸化信号都可能导致疾病的产生。目前,蛋白激酶小分子抑制剂已有许多被批准用于临床治疗,相比之下,蛋白磷酸酶抑制剂的药物研发进展则十分缓慢,大多数进展较快的蛋白磷酸酶抑制剂仍然处于临床实验阶段。变构抑制剂的出现,可以避开高度保守的蛋白磷酸酶催化位点,不仅提高小分子对特定蛋白磷酸酶的选择性,还可以减小分子的极性和电荷,提高溶解性、渗透性和生物利用度,因此为了攻克蛋白磷酸酶抑制剂 “ 不可成药 ” 的难题,蛋白磷酸酶变构抑制剂药物也被寄予厚望。除了本文提到的蛋白磷酸酶,仍有许多蛋白磷酸酶可能存在变构位点且尚未被充分研究,也有一些非竞争性抑制剂的作用位点尚未确证。在本文介绍的变构抑制剂中,人们采用多种方法对蛋白磷酸酶的变构位点进行开发,除了酶动力学分析和酶选择性筛选,NMR、关键残基突变试验、分子对接(docking)以及小分子蛋白质共晶结构解析等诸多方法均有涉及,这些方法也为发现新的蛋白磷酸酶变构位点提供参考。由于开发新位点需要非常多的验证工作,蛋白磷酸酶变构抑制剂的研究往往是机遇与挑战并存,如同十几年前开始研究蛋白激酶药物一样,对蛋白磷酸酶变构抑制剂的研究也许意味着正在打开蛋白磷酸化调控的另一个“宝箱”。
本文整理转载自微信公众号:药学进展(ppsyxjz),作者:药学进展。版权归原作者所有。
学科前沿|生命学院潘俊敏和梁鑫课题组合作在eLife报道马达蛋白Kinesin-II对纤毛长度的调控
2020年10月28日,清华大学生命科学学院潘俊敏和梁鑫课题组合作,在eLife期刊上在线发表了题为“马达蛋白kinesin-II参与“鞭毛内运输”与纤毛长度调控的功能解析” (Functional Exploration of Heterotrimeric Kinesin-II in IF...
本文来源:药学进展 作者:药学进展 免责声明:该文章版权归原作者所有,仅代表作者观点,转载目的在于传递更多信息,并不代表“医药行”认同其观点和对其真实性负责。如涉及作品内容、版权和其他问题,请在30日内与我们联系


 我们沟通的很顺畅
我们沟通的很顺畅 电话已拨通,无人接听
电话已拨通,无人接听 这个电话号码是空号
这个电话号码是空号