 医药魔方Plus
医药魔方Plus基于医药魔方网站行业快讯板块、NextPharma、NextMed数据库以及公开信息,2020年10月的《临床研究月报》共筛选出19项值得关注的临床研究,包括6项肿瘤研究,3项心血管研究、3项COVID-19研究和7项其他领域研究,供您参考。更多行业资讯,点击文末阅读原文,访问医药魔方官方网站。
肿瘤领域
1. Opdivo+Yervoy
10月2日,百时美施贵宝(BMS)公布了Opdivo与Yervoy免疫组合疗法治疗IIIB/C/D期或IV期黑色素瘤的III期临床研究(CheckMate-915)的共同主要终点结果。结果显示,在所有患者(意向性治疗,ITT)中,与Opdivo单药治疗相比,O+Y组合在无复发生存期(RFS)的共同主要终点上,没有达到统计学意义的显著改善。
CheckMate -915是一项随机、双盲、安慰剂对照的III期研究,评估了已完全手术切除的IIIB/C/D期或IV期(无疾病证据)黑色素瘤患者使用Opdivo联合Yervoy与Opdivo单药疗法(已批准的标准治疗)的治疗效果。入组研究的患者除了接受黑素瘤病灶手术和/或中枢神经系统病灶的神经外科切除术后的辅助放射治疗外,之前没有接受过黑素瘤的抗癌治疗。该研究中,共有1934例患者进行了随机分配,接受O+Y组合疗法(Opdivo 240mg,每两周静脉输注一次;Yervoy 1mg/kg,每六周静脉输注一次)或Opdivo单药治疗(480mg,每4周静脉输注一次),治疗时间为一年。此前,2019年11月,BMS曾宣布,在肿瘤已切除且表达PD-L1<1%的高危黑色素瘤患者中,与Opdivo单药治疗相比,O+Y组合在无复发生存期(RFS)的共同主要终点没有达到统计学意义的显著改善。
虽然试验取得阴性结果,但是CheckMate-915结果强化了Opdivo单药在辅助治疗中作为标准护理疗法的既定益处。BMS将完成对CheckMate-915数据的全面评估,并与研究人员在即将召开的医学会议上分享结果。
2. Sotorasib(AMG 510)
10月5日,安进宣布其KRAS G12C抑制剂sotorasib(AMG 510)治疗126例晚期非小细胞肺癌(NSCLC)患者的II期CodeBreaK 100临床试验顶线结果呈阳性,这些患者此前接受过免疫治疗和/或化疗。
研究结果表明,Sotorasib的客观应答率与先前报道的 I 期临床(960mg/日剂量组治疗晚期NSCLC)数据一致,到达主要终点。其他包括应答持续时间在内的疗效指标也令人鼓舞,数据截止时有超过一半的应答患者仍在接受治疗且持续应答。安全性与耐受性与既往研究结果一致。这项潜在性II期注册性临床研究数据将在2021年1月举办的世界肺癌大会中进行公布。
3. Opdivo+化疗
10月7日,BMS宣布,在可切除的NSCLC患者中进行Opdivo联合化疗的III期临床研究(CheckMate-816)达到病理完全缓解(pCR)的主要研究终点。研究结果显示,在术前接受纳武利尤单抗联合化疗的患者中,手术切除标本未发现癌细胞的患者人数显著多于单用化疗的患者。CheckMate-816成为首个,也是目前唯一一个证实免疫检查点抑制剂联合化疗作为新辅助治疗能够为非转移性非小细胞肺癌患者带来获益的III期临床研究。
Checkmate-816是一项随机、开放标签、多中心的III期临床研究,旨在评估与单用化疗相比,纳武利尤单抗联合化疗用于可切除非小细胞肺癌患者新辅助治疗的疗效。在主要分析中,358例患者在术前随机接受纳武利尤单抗(360 mg)联合基于组织学分型的含铂双药化疗(每3周一次,最多3个周期),或者单用含铂双药化疗(每3周一次,最多3个周期)。主要研究终点是病理完全缓解(pCR)和无事件生存期(EFS),关键次要终点包括总生存期(OS)、主要病理缓解(MPR),以及至死亡或远处转移的时间。
BMS将完成对CheckMate-816研究现有数据的全面评估,与研究者共同在即将举行的医学会议上公布研究结果,并将与卫生监管部门讨论潜在的注册路径。CheckMate-816研究目前仍在进行中,以评估另一个主要研究终点无事件生存期(EFS)以及关键次要终点,目前此部分数据对公司依然为盲态。
迄今为止,欧狄沃作为新辅助或辅助治疗已在四种肿瘤类型中显示出了疗效改善,包括肺癌、膀胱癌、食管癌/胃食管连接部癌和黑色素瘤。
4. Ibrance(palbociclib)
10月9日,辉瑞宣布其CDK4/6抑制剂Ibrance(palbociclib)在治疗HR+、HER2-女性早期乳腺癌患者的III期PENELOPE-B研究中未到达无浸润性疾病生存期 (iDFS)的主要终点,这些患者在完成新辅助化疗后仍有残留浸润性疾病。
PENELOPE-B研究是一项随机、双盲、安慰剂对照的III期临床试验,共纳入1250例患者,旨在比较Ibrance联合内分泌标准治疗较安慰剂联合内分泌标准治疗的疗效和安全性。这些患者临床病理分期-雌激素/分级(CPS-EG)评分为3分或更高 (如果手术时有淋巴结转移,则为2分)。研究未观察到新的安全性信号。详细试验数据将在之后进行的医学会议上公布。
这也是Ibrance(palbociclib)今年5月在联合内分泌疗法治疗HR+/HER2-的III期临床研究PALLAS遭遇失败之后,在早期乳腺癌临床研究的又一次失利。而此次PENELOPE-B的临床试验在早期乳腺癌治疗中未达到主要终点,亦没有观察到意外的安全信号。辉瑞方面表示,尽管对这一结果感到失望,仍然希望继续与研究合作伙伴合作,以了解这些数据如何为早期乳腺癌下一代CDK抑制剂的开发提供信息。
5. SGI-110
10月14日,Astex和Otsuka公布了Guadecitabine(SGI-110)的III期试验ASTRAL-2(NCT02920008)和ASTRAL-3(NCT02907359)顶线数据,用于既往接受过治疗的成人患者治疗急性髓性白血病(AML)和骨髓增生异常综合征或慢性粒单核细胞白血病(MDS/CMML)。研究结果显示,SGI-110对比医生选择的替代疗法未显著改善OS,两项试验均未达到主要终点。目前正在评估研究的预期亚组和次要终点,完整数据将在科学会议上呈现。
Guadecitabine是下一代DNA甲基化剂,经过合理设计,可抵抗胞苷脱氨酶降解,从而延长肿瘤细胞对活性代谢产物地西他滨的暴露时间,通过地西他滨的作用抑制DNA甲基转移酶(DNMT),具有逆转异常DNA甲基化的潜能。DNA甲基化是许多癌细胞的表观遗传学特征,并可能恢复沉默的抑癌基因和肿瘤相关抗原的表达,通过这种沉默基因的重新表达,Guadecitabine可能具有使肿瘤细胞对其他抗癌药(包括免疫治疗药物)的敏感性,以及使先前对化学治疗有抗性的癌细胞致敏的潜力。
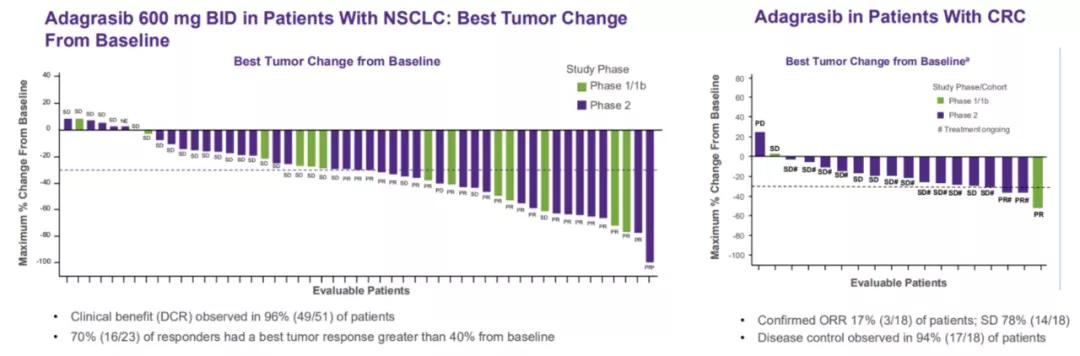
6. Adagrasib(MRTX849)
10月25日,Mirati Therapeutics在第32届国际分子靶标与癌症治疗学研讨会(EORTC-NCI-AACR)上发表KRAS G12C选择性抑制剂adagrasib(MRTX849)在携带KRAS G12C突变的晚期/转移性非小细胞肺癌和结直肠癌中的最新临床数据。
结果显示,在晚期/转移性非小细胞肺癌中,45%的患者对adagrasib(MRTX849)治疗具有客观反应,70%的患者应答者对肿瘤的最佳应答大于40%,患者的疾病控制率为96%。在可评估的CRC患者中,17%(3/18)有确定的客观反应,并且3例患者中有2例仍在接受治疗。94%(17/18)的患者中观察到疾病控制,并且仍有12例患者仍在接受治疗,55%(10/18)的患者治疗时间超过4个月。更多详细内容可参阅此前报道《KRAS抑制剂对比:MRTX849比AMG510更好吗?》。
心血管领域
1. Omecamtiv mecarbil
10月08日,安进与其合作伙伴Cytokinetics和施维雅(Servier)共同宣布了omecamtiv mecarbil在射血分数降低(HFrEF)的心力衰竭患者中进行的关键III期临床研究(GALACTIC-HF)顶线结果。结果显示,omecamtiv mecarbil达到了与安慰剂相比,使心血管(CV)死亡或心衰事件(心衰住院或其他心衰紧急治疗)在统计学上显著减少(HR=0.92;95%CI:0.86,0.99;p=0.0252)的主要复合疗效终点,没有观察到心血管死亡的次要终点降低。不良事件(包括重大缺血性心脏事件)在各治疗组之间未显示差异。GALACTIC-HF的结果将在2020年11月13日美国心脏协会(AHA)的科学会议上进行介绍。
GALACTIC-HF(通过改善心力衰竭的收缩力降低不良心脏结局的全球方案)是迄今为止在心衰治疗领域开展的最大规模III期全球性心血管结局研究之一,在35个国家入组了8256例HFrEF患者,这些患者纽约心脏协会(NYHA)评级为II-IV级、左室射血分数(LVEF)≤35%、利钠肽升高,且在入组研究时因心衰住院,或在筛查前一年内因心衰住院或进入急诊室。研究的目的是评估在标准护理中加入omecamtiv mecarbil治疗是否能降低HFrEF患者发生心衰事件(心衰住院治疗和其他心衰紧急治疗)和心血管(CV)死亡的风险。将患者随机分为口服安慰剂或25 mg omecamtivmecarbil起始剂量,每天两次(维持剂量分别为50 mg,37.5mg或25 mg,每天两次)。
Omecamtiv mecarbil是一种研究性的选择性新型心肌肌球蛋白激活剂,是首个新型肌营养剂。临床前研究表明,心肌肌球蛋白激活剂可在不影响心肌细胞胞内钙浓度或心肌耗氧量的情况下增加心肌收缩力。心肌肌球蛋白是心肌细胞中的细胞骨架运动蛋白,直接负责将化学能转化为导致心肌收缩的机械力。
2. Jardiance(empagliflozin)
10月23日,勃林格殷格翰和礼来公司发布公告,在EMPEROR-Reduced III期临床试验新的探索性亚组分析中表明,恩格列净可降低伴或不伴糖尿病、射血分数降低的成年人心力衰竭患者的心血管和肾脏不良事件的风险,无论其基础性慢性肾脏疾病情况如何。
如先前报道,EMPEROR-Reduced显示Jardiance将患有或未患有糖尿病的心力衰竭伴有射血分数降低的成年人的心血管死亡或因心力衰竭住院的综合终点的相对风险降低了25%,将因心力衰竭而首次住院和反复住院的相对风险降低了30%,并减缓了eGFR(衡量肾功能下降的指标)的下降。另一项探索性分析表明,Jardiance将复合肾终点(包括终末期肾脏疾病和肾功能严重丧失)的相对风险降低了50%。在这项新分析中的所有终点中,在基线时有或没有慢性肾脏病的患者(包括严重肾功能不全的患者)的亚组中均观察到了这些益处。在参加EMPEROR-Reduced试验的所有患者队列中,安全性与Jardiance已知的安全性相似。
3. Forxiga(dapagliflozin)
10月,国家药品监督管理局(NMPA)批准更新阿斯利康安达唐(达格列净,dapagliflozin)中国说明书,纳入了DECLARE-TIMI 58 III期临床研究结果中的部分数据。
DECLARE-TIMI 58研究是一项由阿斯利康申办的随机、双盲、安慰剂对照、多中心的 III期临床研究,旨在评估达格列净与安慰剂相比,对具有心血管事件风险(包括多重心血管危险因素或已确诊的心血管疾病)的2型糖尿病(T2D)成年患者的心血管结局,同时也评估了关键肾脏次要终点。DECLARE-TIMI 58研究纳入了来自33个国家、882个中心的17,000多例患者,由TIMI研究组(马萨诸塞州,波士顿)独立运行,并与哈达萨希伯来大学医学中心(以色列,耶路撒冷)合作。
DECLARE-TIMI 58研究显示:与安慰剂相比,达格列净能够有效降低已确诊心血管疾病或多重心血管危险因素的成人2型糖尿病患者因心衰住院(hHF)或心血管死亡的风险。该研究所示安全性与达格列净已知安全性一致。
COVID-19
1. LY-CoV555+LY-CoV016
10月7日,礼来宣布了其抗SARS-CoV-2病毒中和抗体开发项目的最新进展,包括两款中和抗体 LY-CoV555与LY-CoV016(从君实引进的JS016)的联合疗法用于新确诊轻至中度COVID-19患者的BLAZE-1研究最新期中数据。
BLAZE-1研究(NCT04427501)为一项随机、双盲、安慰剂对照、II期研究,主要评估LY-CoV555和LY-CoV016相比安慰剂用于治疗伴有症状COVID-19门诊患者的疗效及安全性差异,计划入选800例症状轻至中度且药物输注前3日内SARS-CoV-2样本检测为阳性的COVID-19患者。
该研究的单药治疗队列评估LY-CoV555三个剂量 (700,2800,7000mg)的治疗效果和安全性,联合用药组评估LY-CoV555(2800mg)+LY-CoV016(2800mg)的治疗效果和安全性。安慰剂组患者在完成各队列研究后允许进入全部受试药组交叉用药。研究主要终点是第11天SARS-CoV-2 病毒载量较基线的变化。其他终点包括因COVID-19住院的患者比例,29天内死亡率、安全性等。
截至期中分析时,联合疗法组入组112人,安慰剂对照组入组156人。结果显示,LY-CoV555+ LY-CoV016联合疗法显著降低了第11天的病毒载量(p=0.011),达到了该研究的主要终点。联合疗法还降低了第3天(p=0.016)及第7天(p<0.001)(感染过程中较早的时间点,通常能观察到更高的病毒载量)的病毒水平。该联合疗法亦达到预设的临床终点,包括自第1至11天的症状总体评分相较于基线的时间加权平均变化。探索性分析结果显示,联合疗法组第7天病毒载量依然较高的患者比例低于安慰剂对照组(3% vs 20.8%,p<0.0001)。联合疗法组COVID-19相关住院和急诊治疗的发生率低于安慰剂对照组(0.9% vs 5.8%),风险降低84.5%。
2. Merimepodib+ Remdesivir
10月26日,BioSig Technologies及其控股子公司ViralClearPharmaceuticals宣布停止Merimepodib联用瑞德西韦(Remdesivir)在晚期冠状病毒病(COVID-19)中的临床开发。
该决定是基于一项随机、双盲、安慰剂对照的II期研究,评价口服Merimepodib与静脉注射瑞德西韦联用治疗成人冠状病毒性疾病患者(COVID- 19)的疗效。实施方案修订后,将试验的规模从40位住院的COVID-19患者扩大到80位,并且仅限于重症患者入组(NIAID 3级,需要高流量、高浓度的氧气)。安全监督委员会(SMC)在对数据进行最新审查时,已有44例患者入组,其中42例接受了研究药物(Merimepodib或安慰剂)。
数据的最新审查结果表明,所有22位4级患者均已出院,并且在37天的随访期内未复发。但是,入组时为NIAID 3级患者(n=20)的患者结局明显不同,具体而言,无盲SMC监测到这些NIAID 3级患者的生存率在安慰剂和merimepodib之间的不平衡(imbalance in survivalrates),因此该试验不太可能达到其主要安全性终点。因此,该公司选择停止注册该临床试验。将按照方案对患者进行安全监控;但是,将不会进行进一步的药物研究治疗。
3. REGN-COV2
10月28日,再生元(Regeneron)宣布,其新冠中和抗体鸡尾酒疗法REGN-COV2在一项正在进行的II/III期临床试验中达到主要和关键性次要终点。REGN-COV2显著降低患者的病毒载量和接受进一步医疗护理的需求(包括住院、急诊室就诊、紧急护理和/或医生办公室/远程医疗访视)。
这项随机、双盲试验正在评估将REGN-COV2加入到常规护理标准中与将安慰剂加护理标准中相比的效果。先前报道了对前275名患者的描述性分析,此次报告的数据涉及另外524名患者。
结果显示,在病毒学结果方面,接受治疗7天后,新增患者(n=524)的平均病毒载量与对照组相比降低0.36个log10(p=0.0003,1个log10为10倍)。在基线病毒载量更高的患者中(定义为病毒载量超过1X107拷贝/毫升),REGN-COV2与安慰剂组相比将病毒载量降低0.68个log10(p<0.0001)。在接受治疗后第5天,REGN-COV2与对照组相比,将病毒载量降低1.08个log10,意味着这些患者的病毒载量与对照组相比降低10倍以上。在关键的临床终点上,在第29天使用REGN-COV2治疗可使COVID-19相关的医疗就诊次数减少了57%(2.8%联合剂量组;6.5%安慰剂;p = 0.024)。在具有一种或多种危险因素(包括50岁以上;体重指数大于30;心血管、代谢、肺、肝或肾疾病;包括糖尿病或免疫功能低下)的患者中,使用REGN-COV2治疗可将COVID-19相关的医疗就诊次数减少72%。
结果还显示,高剂量(8克)和低剂量(2.4克)之间的REGN-COV2病毒学或临床疗效无明显差异。基于这一发现,鉴于目前REGN-COV2的供应有限,Regeneron正在审查正在进行的门诊临床试验中剂量的潜在变化,并计划寻求紧急授权使用。
然而,10月30日,再生元再次发布公告,由于存在潜在的安全隐患,已暂停了用于治疗重症新冠患者抗体药物的临床研究。再生元公司表示,针对新冠病毒的抗体鸡尾酒疗法试验的独立数据监测委员会(IDMC)建议“修改”患者试验,因此部分患者的招募计划被搁置。独立数据监测委员会(IDMC)在一份声明中表示,建议在进一步的数据分析前,暂停进一步招募需要大量吸氧或呼吸机的患者,但是可以继续招募不需要吸氧或只需要少量吸氧的轻症患者,并继续在不加修改的情况下进行门诊试验。
其他
1. PXL770
10月01日,POXEL SA发布公告称,PXL770用于NASH治疗的IIa期临床试验达到了其主要疗效终点,患者在接受12周治疗时,肝脂肪相对减少方面具有统计学上的显著改善,在2型糖尿病患者中效果更加显著。在接受PXL770治疗的患者中,主要的次要措施包括肝酶-丙氨酸转氨酶(ALT)和血红蛋白A1c(HbA1c)的统计学改善,并且PXL770安全且耐受性良好。
这也是直接AMPK激活剂的首次人类临床评估,结果支持包括主要高风险亚组(2型糖尿病患者)在内的NASH治疗潜力,以及AMPK激活剂在其他慢性和罕见代谢疾病中的应用。
2. Zeposia(ozanimod)
10月10日,BMS宣布了True North研究的详细结果。True North是一项关键、安慰剂对照的III期试验,评估口服Zeposia(ozanimod)作为中度至重度溃疡性结肠炎(UC)成年患者的诱导和维持疗法。True North达到了两个主要终点,在第10周诱导期(18.4% vs 6.0%;p<0.0001)和52周维持期(37.0%vs 18.5%,p <0.0001),与安慰剂相比,具有较高的统计学意义和临床意义。该研究还达到了关键的次要终点,包括临床反应、内窥镜改善和第10周诱导时的黏膜愈合以及第52周维持情况。与安慰剂相比,Zeposia治疗的患者在第10周达到临床反应的比例明显更高(47.8% vs 25.9%;p<0.0001)和第52周时(60.0% vs 41.0%;p <0.0001)获得临床疗效的患者明显多于安慰剂组,且各子分析结果一致。观察到的总体安全性与Zeposia和中度至重度UC患者的已知安全性一致。
3. Filgotinib
10月12日,吉利德和GalapagosNV更新了Filgotinib(口服、每天一次的JAK1抑制剂)用于治疗中度至重度活动性溃疡性结肠炎(UC)的最新数据,证明了其持续的疗效和安全性。来自随机、双盲、安慰剂对照的IIb/III期SELECTION试验数据表明,与安慰剂相比,200 mg filgotinib治疗的患者在第10周达到临床缓解并在58周之前保持缓解的比例显著更高。此外,显著更多的患者实现了六个月无皮质类固醇的缓解。
基于该数据,11月02日,EMA批准Filgotinib的新适应症申请,用于治疗对常规疗法或生物制剂的反应不足、反应迟缓或不耐受的中度至重度活动性溃疡性结肠炎(UC)的成年患者。
4. Dupixent
10月13日,赛诺菲宣布其Dupixent(dupilumab)在治疗6-11岁儿童中重度哮喘的关键性III期临床试验中到达主要终点和关键的次要终点。Dupixent也是目前唯一一个在随机III期研究中证明能改善儿童肺功能的生物制剂。
这项代号为LIBERTY ASTHMAVOYAGE的随机、双盲、安慰剂对照的III期临床试验,旨在评估Dupixent联合标准疗法对于儿童中重度哮喘患者的疗效和全性。主要终点为评估2个预先指定人群的重度哮喘发作年化率:嗜酸性粒细胞(EOS)≥300 cells/μl和具有2型炎症标志物(FeNO ≥20 ppbor EOS ≥150 cells/μl)患者。结果显示,接受Dupixent治疗的患者较安慰剂组重度哮喘年化率分别降低了65%和59%。与基线期相比,Dupixent治疗组患者肺功能分别改善了10.15和10.53个百分点,而安慰剂组为4.83和5.32个百分点。安全性数据与既往结果一致。
5. AMX0035
10月16日,Amylyx公布了AMX0035治疗肌萎缩侧索硬化症的II/III期试验(CENTAUR)数据。研究结果显示,随访期间,AMX0035相比安慰剂降低死亡风险达44%(HR,0.56;95%CI,0.34-0.92;P=0.023)。此前披露数据显示,AMX0035 vs. 安慰剂的ALSFRS-R得分平均变化率为-1.24 vs -1.66。这是首个同时显示长期生存获益和功能改善的治疗ALS试验。
AMX0035是一种研究性神经保护疗法,旨在通过靶向线粒体和内质网相关的细胞死亡途径来减少运动神经元的死亡和功能障碍。这些途径代表了与目前批准的药物利鲁唑(Riluzole)和依达拉奉(Edaravone)不同。大多数CENTAUR参与者(77%)在试验进入期间和/或之前接受了批准的ALS治疗(利鲁唑、依达拉奉或两者兼有),并被允许在OLE期间继续使用这些药物。生存分析比较了最初随机分配给AMX0035的参与者和最初随机分配给安慰剂的参与者之间的死亡时间(全因死亡率),在随机分组后的35个月中,在随访过程中,死亡风险降低了44%,AMX0035的生存获益与利鲁唑和/或依达拉奉或两者并用的基线使用无关。
6. SHR0302
10月26日,瑞石生物医药(瑞石生物)宣布,其在研的国内首创、具有自主知识产权的高选择性JAK1抑制剂SHR0302用于治疗特应性皮炎的II期临床研究(QUARTZ2)获得成功,达到试验主要及次要终点。
这项随机、双盲、安慰剂对照、多中心临床研究在中国23个研究中心进行。纳入研究的105名成人受试者均为既往疾病控制不佳的患者,被随机分配每日口服8mg或4mg的SHR0302片或安慰剂,疗程为12周。主要终点为第12周时达到研究者总体评分(IGA)应答的受试者百分比。研究结果显示,两种治疗剂量均明显改善了患者的临床症状,并提高了生活质量。患者在接受了8mg或4mg SHR0302片单一疗法后,皮肤病灶清除显著有效。12周时,接受8mg治疗的患者中54.3%获得IGA应答,接受4mg治疗的患者中25.7%达到IGA应答(8mg vs 安慰剂,p<0.001;4mg vs 安慰剂,p=0.022)。安全性方面,SHR0302总体耐受性良好。试验中没有死亡或危及生命的病例报告,没有严重感染的病例报告,也没有恶性肿瘤或血栓形成的情况。
7. Upadacitinib
10月29日,在第29届欧洲皮肤病和性病学会(EADV)虚拟大会上,艾伯维(Abbvie)公布了来自Measure Up 1和2的upadacitinib单药疗法研究(RINVOQ)的新分析数据,与安慰剂相比,使用upadacitinib(15 mg或30 mg;每天一次)治疗的皮炎患者在皮肤清除率、瘙痒和生活质量的其他衡量指标有所改善,没有发现新的安全风险。
数据来自III期Measure Up 1和Measure Up 2研究,结果支持最近向美国食品药品监督管理局和欧洲药品管理局提交的申请,寻求批准upadacitinib在成人和青少年中度至重度特应性皮炎患者中的使用批准。在MeasureUp 1和2两项研究中,在第16周,较接受安慰剂治疗的患者相比,接受两种剂量的upadacitinib治疗的患者的湿疹面积严重性指数(EASI 90)至少改善了90%以上(p<0.001);此外,对于两种剂量的upadacitinib,在第4周时达到临床意义(NRS≥4.1)的瘙痒减轻患者比例显著高于安慰剂,并维持到第16周。今年早些时候,AbbVie公布了MeasureUp 1和Measure 2研究的主要数据,显示upadacitinib(15 mg或30 mg)达到了共同主要终点和所有次要终点(p <0.001)。
本文源自头条号:医药魔方,作者:。版权归原作者所有。
加强版PI3K抑制剂来了!高效抑制小鼠肺纤维化,提高生存率
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种致命的肺部疾病,好发于中老年人。由于胶原蛋白过度沉积,肺实质逐渐变硬变厚,导致肺部的气体交换功能逐渐失效,患者的预期寿命仅为3-5年。虽然FDA已经批准了两种治疗特发性肺纤维化的药物(吡非...
本文来源:医药魔方Plus 作者:小编 免责声明:该文章版权归原作者所有,仅代表作者观点,转载目的在于传递更多信息,并不代表“医药行”认同其观点和对其真实性负责。如涉及作品内容、版权和其他问题,请在30日内与我们联系


 我们沟通的很顺畅
我们沟通的很顺畅 电话已拨通,无人接听
电话已拨通,无人接听 这个电话号码是空号
这个电话号码是空号